
揭秘“隐秘角落”的病毒风险:华大等团队揭示中国西南食虫动物病毒组图谱
2025年5月16日,华大火眼联合军事医学科学院、安徽医科大学等团队在国际顶刊 《Microbiome》 发表重要研究成果。该研究首次对中国西南生物多样性热点地区的食虫动物(鼩鼱、刺猬、鼹鼠)进行了系统性的病毒组学分析,为理解这些“隐秘”宿主的病毒多样性、生态驱动因素及人畜共患病风险提供了关键数据。
核心研究发现
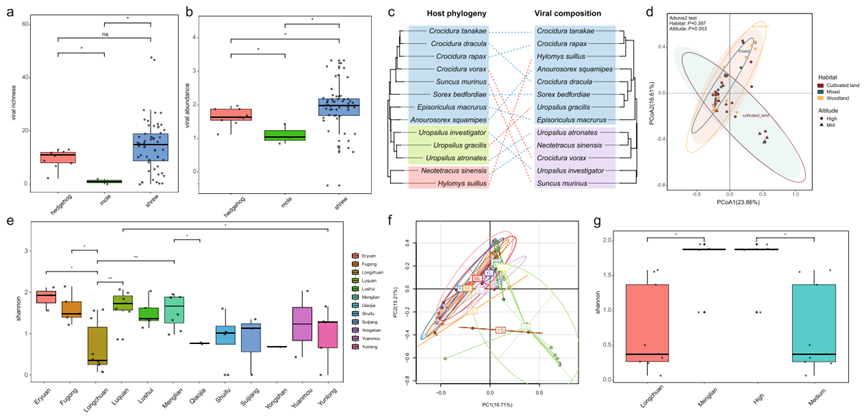
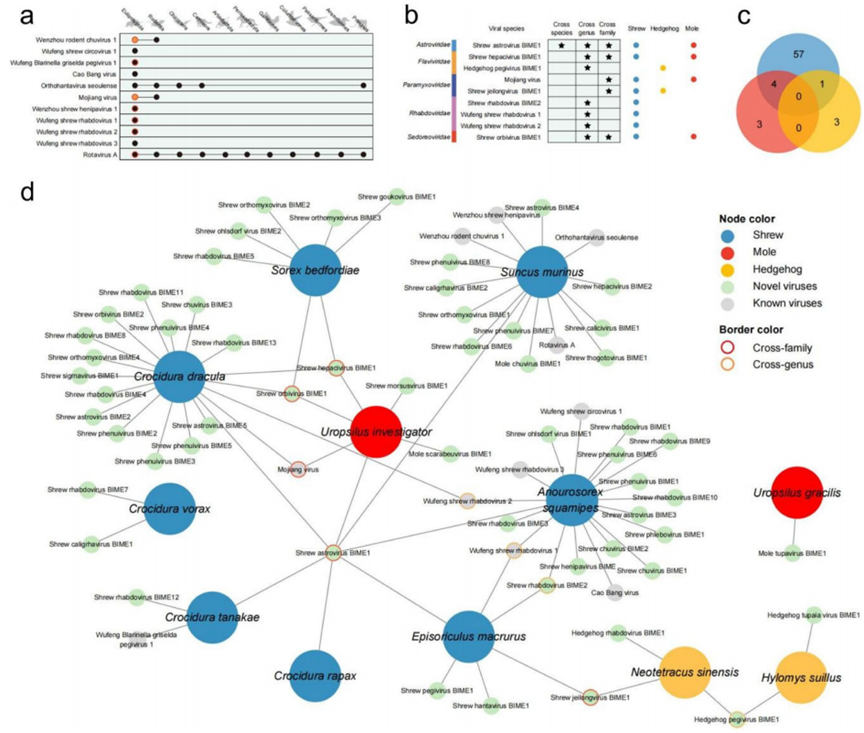
- 海量病毒数据: 从214只食虫动物中鉴定出42个病毒科,发现了57种前所未见的新病毒。
- 宿主差异显著: 鼩鼱携带的病毒多样性和丰度显著高于刺猬和鼹鼠,是多种病毒的重要宿主。
- 生态驱动病毒分布: 病毒的组成受到宿主分类、海拔和地理位置的显著影响。
- 揭示跨种传播: 发现了11种病毒存在跨宿主传播现象,尤其是在鼩鼱和鼹鼠之间,其中鼹鼠可能扮演了关键的“中介”角色。
- 潜在公共卫生风险: 在样本中检测到与墨江副黏病毒和汉坦病毒等已知病原体同源性极高的病毒序列,提示存在向人类传播的潜在风险。
研究亮点与方法学启示
本研究不仅提供了宝贵的病毒生态数据,其系统性的研究框架也为未来的病原体监测提供了重要参考。
-
聚焦“核心病毒组”
选择宿主器官(肺组织)而非环境样本(粪便)进行分析,能更准确地反映宿主真实感染的病毒谱,有效排除了环境污染的干扰。 -
多维度生态因子建模
综合宿主分类、地理分布、海拔、栖息地等多重因素,系统解析了影响病毒组差异的关键驱动力,为地理医学研究提供了范例。 -
构建跨物种传播网络
利用生物信息学工具直观地展示了病毒在不同物种间的传播路径,并成功识别出鼹鼠这一潜在的关键“桥梁”物种。 -
从发现到预警的转化
研究不仅止步于发现新病毒,更基于病毒丰度、宿主分布和传播风险,为建立长期、主动的公共卫生预警监测网络提供了科学依据。
结论
这项研究系统地揭示了中国西南地区食虫动物这一长期被忽视的病毒库,强调了将宿主、环境与病毒进行整合分析的重要性。研究成果不仅极大地丰富了我们对病毒生态学的认知,也为全球人畜共患病的早期预警和防控策略提供了新的视角和关键数据支持。
研究方法详解
一、实验设计
本研究旨在填补食虫动物病毒组的研究空白。通过对云南省12个县的214只食虫动物(13个物种)的肺组织样本进行宏转录组测序,结合宿主分类、地理和生态数据,系统分析其病毒组多样性、驱动因素及跨物种传播风险。
二、样本采集与处理
- 采集: 2015至2020年间,在云南12个县的自然、人工及混合栖息地(海拔500-3561米)采集样本,并记录详细生态信息。
- 物种鉴定: 通过形态学结合
cytb和COI基因测序进行精准物种鉴定。 - 样本处理: 无菌解剖获取肺组织,提取总RNA,并将来自同一物种和地点的样本合并为68个混合池以进行后续分析。
三、测序与生物信息学分析
- 测序: 构建宏转录组文库,在MGISEQ-2000平台进行高通量测序。
- 病毒鉴定: 采用
Fastp质控、MEGAHIT从头组装、DIAMOND blastx和BLASTn进行序列比对和注释,最终手动验证新病毒。 - 系统发育分析: 使用
MAFFT进行序列比对,IQ-TREE构建最大似然系统发育树。 - 多样性与统计分析:
- α多样性: 计算Shannon指数比较不同宿主群体的病毒丰富度。
- β多样性: 通过PCoA分析和PERMANOVA检验评估不同环境因子对病毒组成的影响。
- 差异检验: 使用Wilcoxon秩和检验和Kruskal-Wallis检验进行组间比较。
文章目录
文章作者:火眼工程
文章标题:火眼工程与中国科学家合作完成中国西南地区食虫动物病毒组及病毒传播风险相关研究
文章链接:https://huoyan.genomics.cn/?post=2
本站所有文章转载请注明来自 华大火眼工程 !
文章标题:火眼工程与中国科学家合作完成中国西南地区食虫动物病毒组及病毒传播风险相关研究
文章链接:https://huoyan.genomics.cn/?post=2
本站所有文章转载请注明来自 华大火眼工程 !
设备上扫码阅读